Realize and Optimize Cell Segmentation in Spatial Transcriptomics
This work has been presented in the Conference of Academic Communication for Talented Programs (基础学科英才班学术交流会)(news report about this event) at University of Science and Technology of China. SlidesSlides in Google Drive.
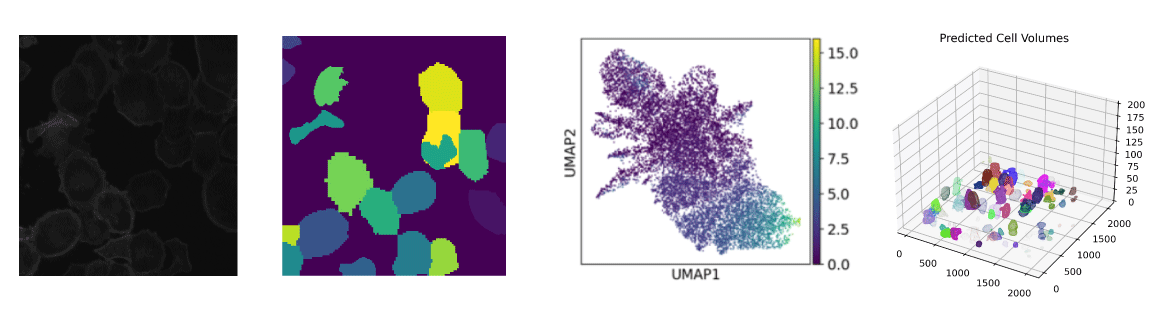
Cell segmentation is an important step in the pre-processing of the spatial transcriptomics data. Nowadays the technologies of spatial transcriptomics can be mainly classified as two types: Based on NGS and based on image. For these methods that using image to detect the signal of molecular (probe), we need to assign each signal (point at the image) to a given cell. The cell boundary can be marked using H&E staining.
After assign the signal to the cell, the data becomes "single cell transcriptomics data with the position". Then the data can be used to analysis using the standard workflow of single-cell analysis.
However, in May. 2021, when I start the project, the cell segmentation tools are not optimal. The most commonly used methods are based on the watershed algorithm, which is not suitable for the tools developed by our lab
Being new in the computational field, I had a lot to learn. But I successfully utitalized three tools based on deep learning to our own dataset. In the process I rewrite and rearrange large part of the source code, which utimately significantly improved the accuracy of cell segmentation in our dataset.
Another problem in the spatial transcriptomics upstream analysis is to make use of the 3D image. The format of the data is OME-TIFF. How to use the image data from different focus plane (z-stacks)? Currently, the most useful method is the maxiumn intensity projection, but it do not use the signal from all z-stacks. With the help of colleagues, we developed an algorithm to track the same segmented cell in different z-stacks. This algorithm has the promise to utilize all information from the image data.